
This example performs molecular Monte Carlo (MC) simulations on B-form double stranded deoxyribonucleic acids (B-DNA) and protein backbones, to generate many configurations of a nucleosome core particle (NCP), a DNA-protein complex.
The directionality for defining a flexible B-DNA region is based on the 5' to 3' direction of the first DNA segname in the flexible region definition.
Care must be taken to correctly include the matched DNA base pairs when defining a flexible region. The code may run when given misaligned input but the results will not be usable.
The starting structure must be a complete structure without missing residues. Atom and residue naming must be compatible with those defined in the CHARMM force field. See the pages on Structures and Force Fields and PDB Scan for further details.
Structures are generated by Markov Monte Carlo sampling of bend and twist moves of B-DNA represented using the bead-rod model based on the worm-like chain and protein backbone torsion angles. Energetics of the B-DNA moves are discussed further in this publication. Energetics of torsion angles are determined using CHARMM force field parameters.
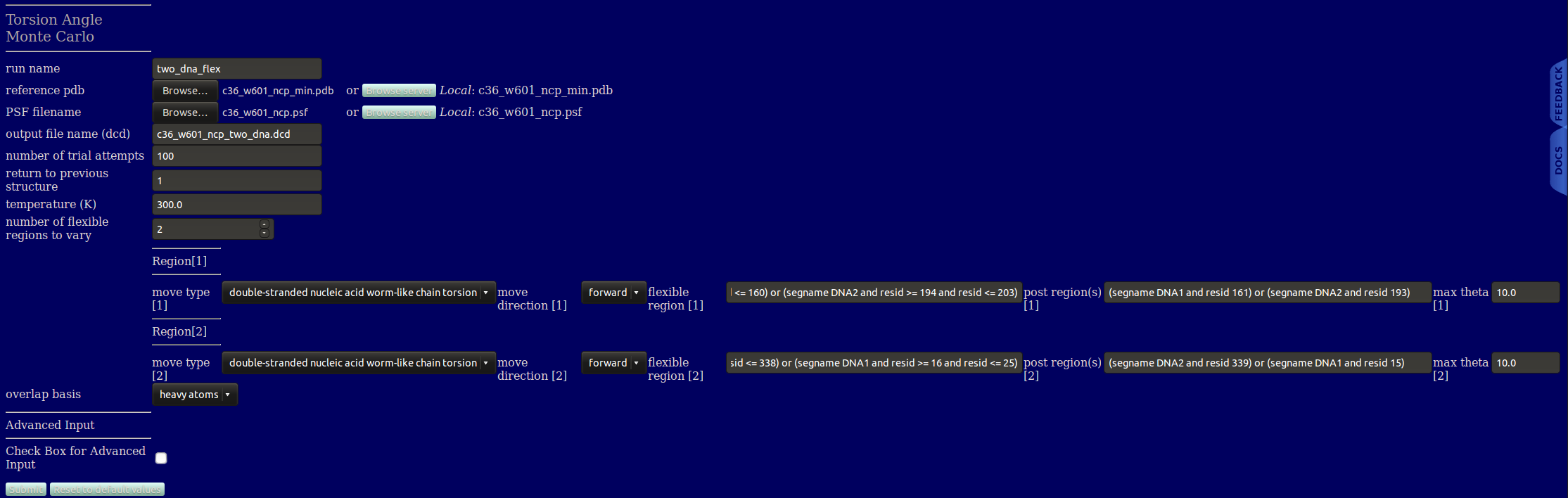
run name user defined folder name for storing the results.
reference pdb PDB file with atomic information and coordinates for the starting structure.
PSF Filename PSF file with topology information, must match the reference PDB file.
output file name Name of output DCD file containing the accepted structures.
number of trial attempts Number of Monte Carlo moves to attempt.
return to previous structure After this number of Monte Carlo moves fails to find an accepted configuration, re-load a previously accepted structure. If this value is set to 1, any failed step will backup to the previous configuration then attempt a new Monte Carlo move. For values greater than 1, a random structure form the previously accepted structures is selected. When the simulated system contains B-DNA, typically users should use a value of 1 to explore large variations from the starting configuration. If you think of the Monte Carlo simulation as a walk with the initial configuration as the starting location, a value of 1 would represent continuously walking away from that starting location, while a value greater than 1 would represent repeatedly returning to locations previously visited. The first option will typically be the quickest method for exploring configurations far from the starting structure, while the later option will more thoroughly explore configurations around the starting point.
temperature (K) Simulation temperature use for calculating energies from CHARMM force field parameters and calculating the screened Debye electrostatic energy.
number of flexible regions to vary An integer value indicating the number of regions to sample torsions. The value entered will dictate the number of further inputs to be created.
move direction This parameter applies to protein backbone, single-stranded nucleic acid (ssNA), and isopeptide bond torsions, but does not apply to B-DNA moves (the page on B-DNA directionality explains how to define the move direction for B-DNA). For protein and ssNA systems, forward and reverse respectively indicate that the moved residues will be those with a resid larger and smaller than those in the flexible region. For isopeptide systems, forward and reverse respectively indicate the moved residues will be those on the lysine and C-terminal sides of the isopeptide bond. The selected option must match the selection defined in the post region input.
flexible region Unique descriptor to define each flexible region (VMD like input).
post region Unique descriptor to define each flexible region (VMD like input).
max theta Maximum angle, in degrees, to sample in a single MC move. This input is specific to each region. The default values for protein and B-DNA are respectively 30 and 10 degrees. These values typically provide both a reasonable amount of both variation and acceptance.
overlap basis Select the basis atoms used in determining overlap. Available options are:
The notation used for defining flexible and post regions follows the notation used in VMD (this does not include shortcuts and generic options such as 'backbone', 'sidechain', 'nucleic', 'water', 'protein', etc). Every flexible B-DNA region should include the paired bases from 2 DNA segments. It is critically important to enter the correct residues for the DNA base pairing; misaligned input will produce nonsense results. As a general rule, do not include the first or last residue of a segment in a flexible region.
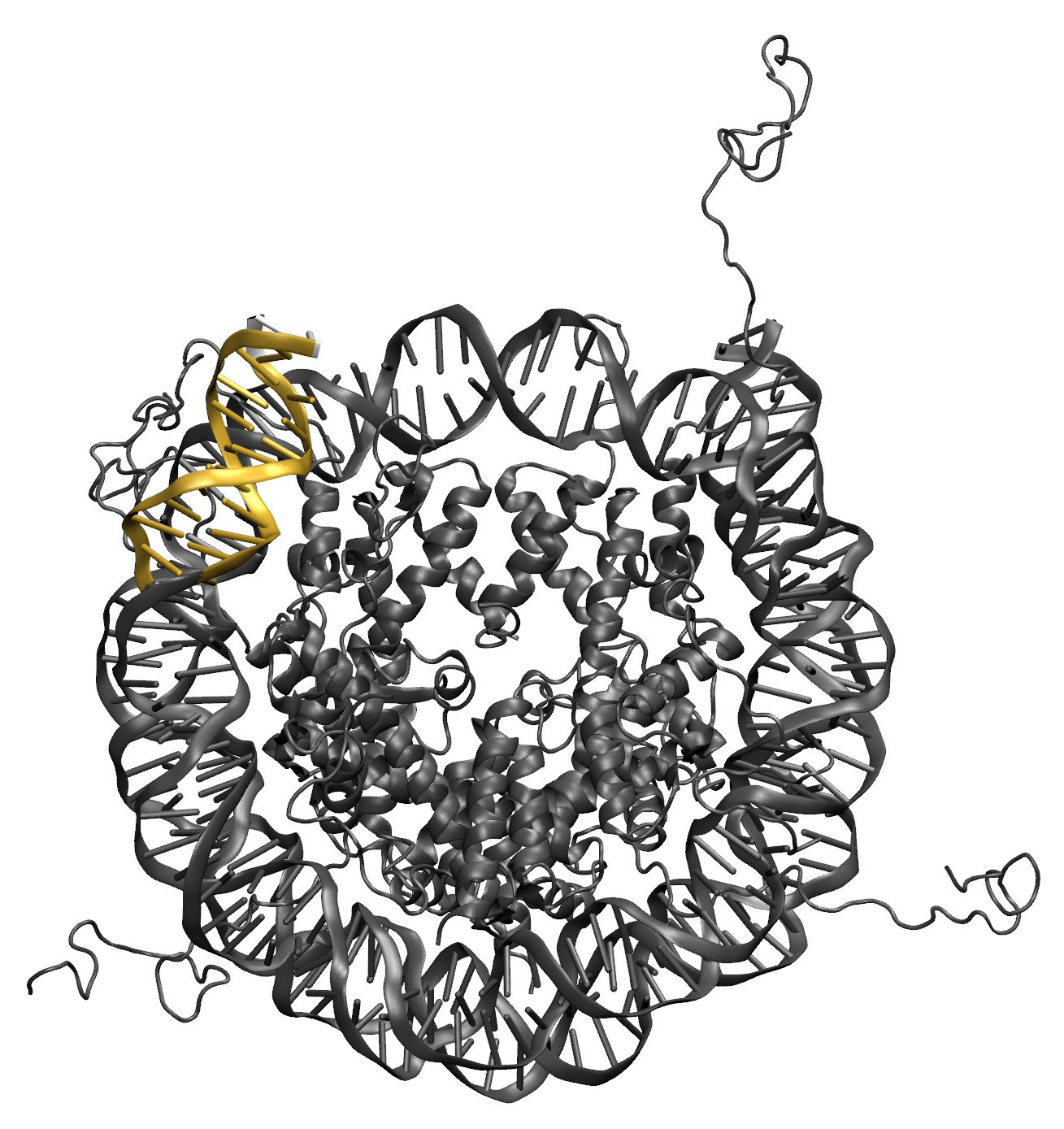
Illustrations of the starting structure highlighting the flexible region in yellow, and the post region in white (note that the directionality used to define the post region follows the 5' to 3' direction of the first DNA segment in the flexible region). Notice that the end base pair (white) is not designated as flexible. This prevent problems in the Monte Carlo Sampling caused by the uniqueness of the end base pairs, which are only connected to one other base pair. The input definitions for this selection is as follows:
move type:
double stranded nucleic acid worm-like chain torsion
flexible region:
(segname DNA1 and resid >= 151 and resid <= 160) or (segname DNA2 and resid >= 194 and resid <= 203)
post region:
(segname DNA1 and resid 161) or (segname DNA2 and resid 193)
max theta:
10.0
The output will indicate the location of the output files, acceptance and overlap statistics, and the file names of the inputs, log, and output DCD. Results are written to a new directory within the given "run name" as noted in the output. In addition, a plot of Rg versus structure number is shown (currently NOT implemented).
Several files are generated and saved to the <run name>/monte_carlo/ directory: a copy of the original input PDB and PSF files, the output DCD file containing accepted structures, a PDB and PSF file for each group, flexible region, and post region, the json inputs, and a log file. In this example, the dcd containing the generated structures accepted by the Monte Carlo algorithm is one_dna_flex/monte_carlo/c36_w601_ncp_one_dna.dcd.
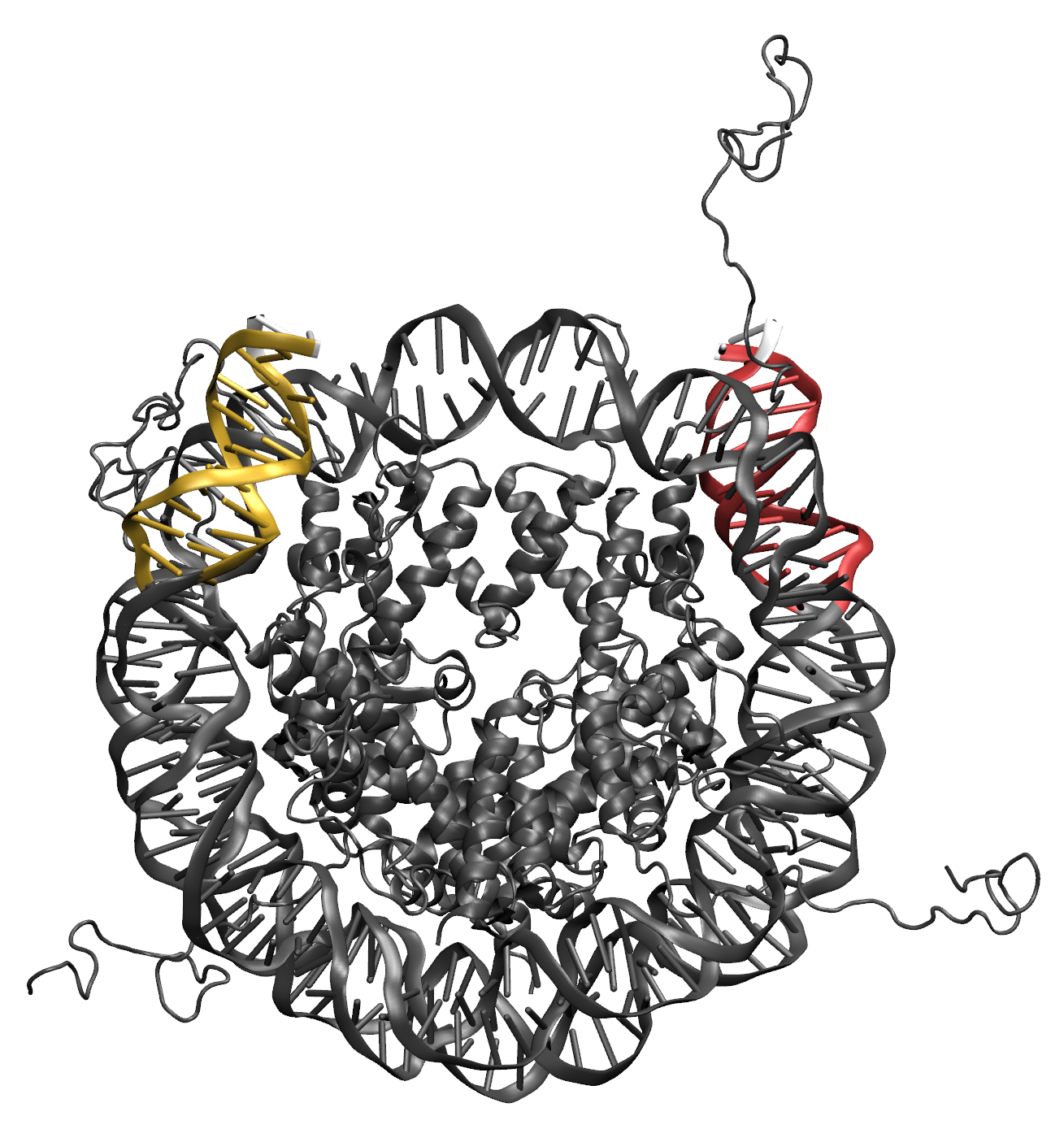
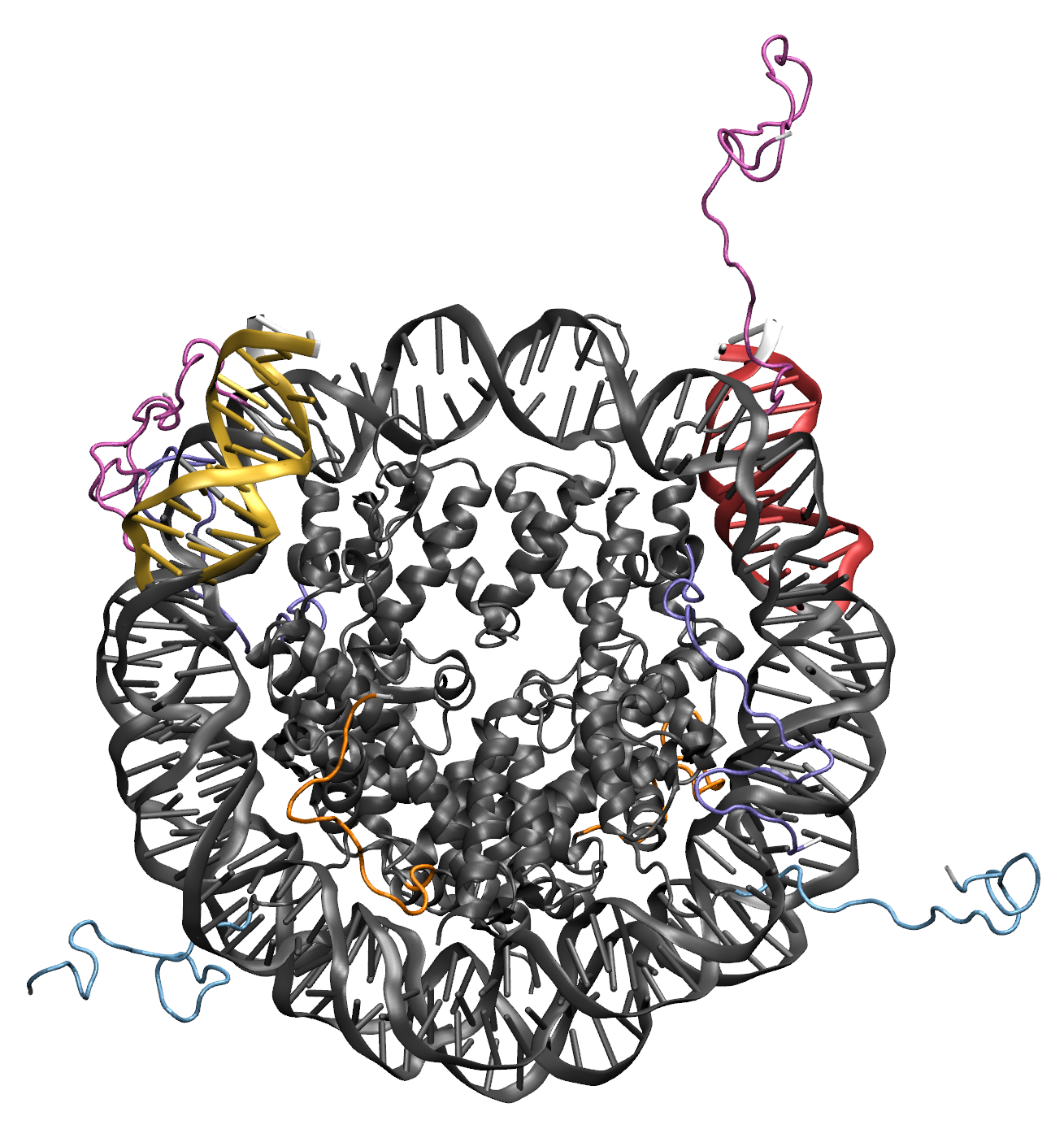
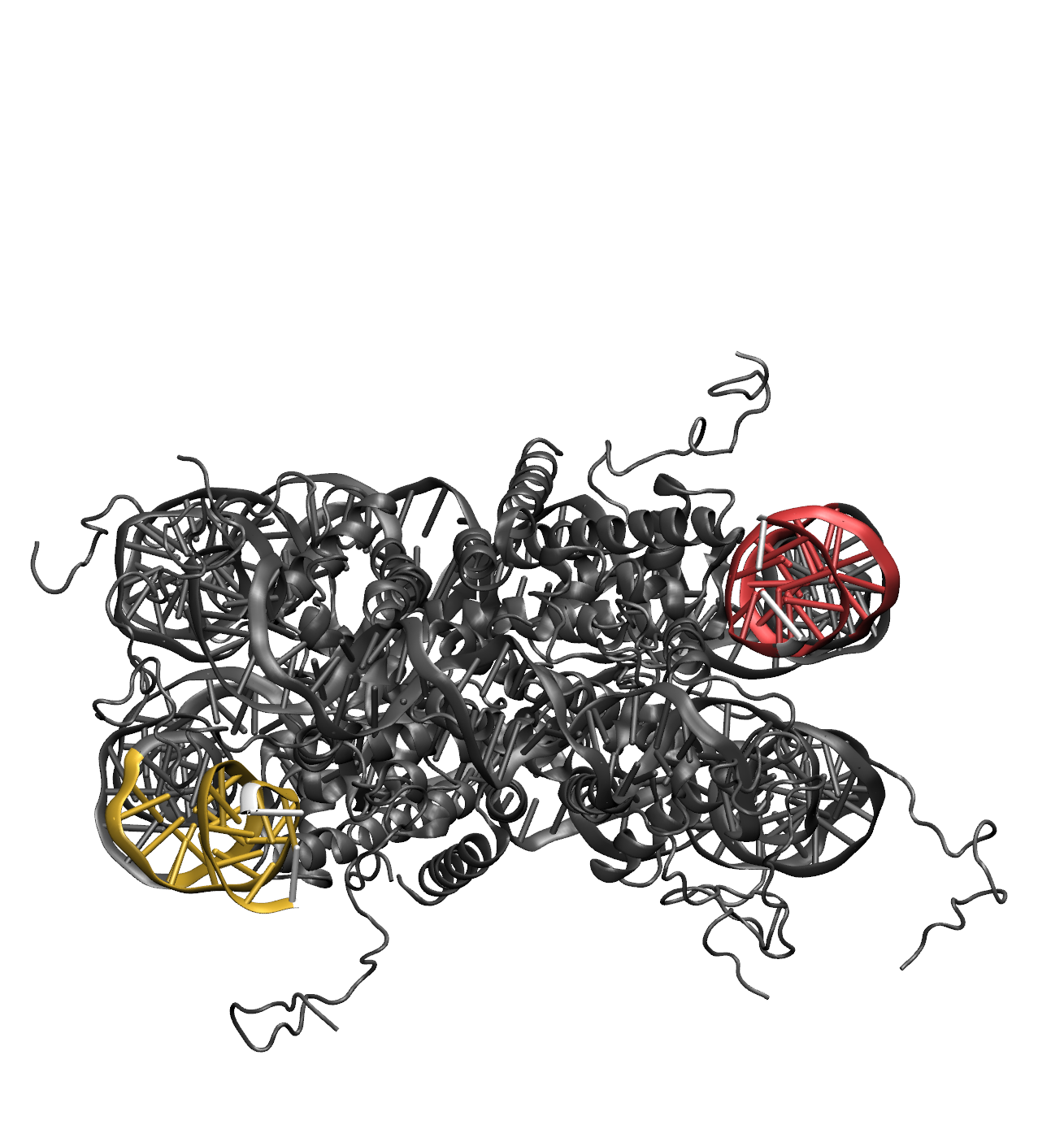 Illustrations of the starting structure highlighting the flexible regions in yellow and red, and the post regions in white (note that the directionality used to define the post region follows the 5' to 3' direction of the first DNA segment in the flexible region). Notice that the end base pairs (white) are not designated as flexible. This prevent problems in the Monte Carlo Sampling caused by the uniqueness of the end base pairs, which are only connected to one other base pair. The input definitions for this selection is as follows:
Illustrations of the starting structure highlighting the flexible regions in yellow and red, and the post regions in white (note that the directionality used to define the post region follows the 5' to 3' direction of the first DNA segment in the flexible region). Notice that the end base pairs (white) are not designated as flexible. This prevent problems in the Monte Carlo Sampling caused by the uniqueness of the end base pairs, which are only connected to one other base pair. The input definitions for this selection is as follows:
move type:
double stranded nucleic acid worm-like chain torsion
double stranded nucleic acid worm-like chain torsion
flexible regions in order:
(segname DNA1 and resid >= 151 and resid <= 160) or (segname DNA2 and resid >= 194 and resid <= 203)
(segname DNA2 and resid >= 329 and resid <= 338) or (segname DNA1 and resid >= 16 and resid <= 25)
post regions in order:
(segname DNA1 and resid 161) or (segname DNA2 and resid 193)
(segname DNA2 and resid 339) or (segname DNA1 and resid 15)
Notice: placing segname DNA2 first in the second flexible region reverses the directionality for defining the post region.
max theta:
10.0
10.0
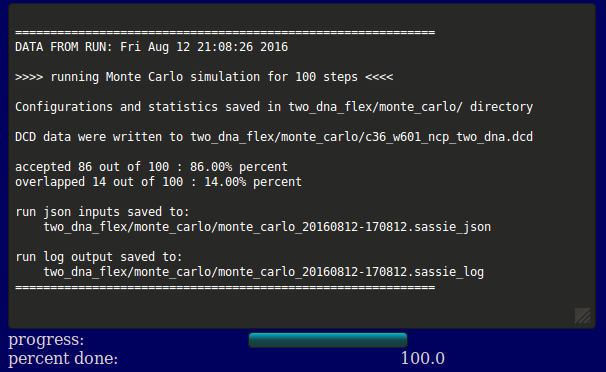
The output will indicate the location of the output files, acceptance and overlap statistics, and the file names of the inputs, log, and output DCD. Results are written to a new directory within the given "run name" as noted in the output. In addition, a plot of Rg versus structure number is shown (currently NOT implemented).
Several files are generated and saved to the <run name>/monte_carlo/ directory: a copy of the original input PDB and PSF files, the output DCD file containing accepted structures, a PDB and PSF file for each group, flexible region, and post region, the json inputs, and a log file. In this example, the dcd containing the generated structures accepted by the Monte Carlo algorithm is two_dna_flex/monte_carlo/c36_w601_ncp_two_dna.dcd.
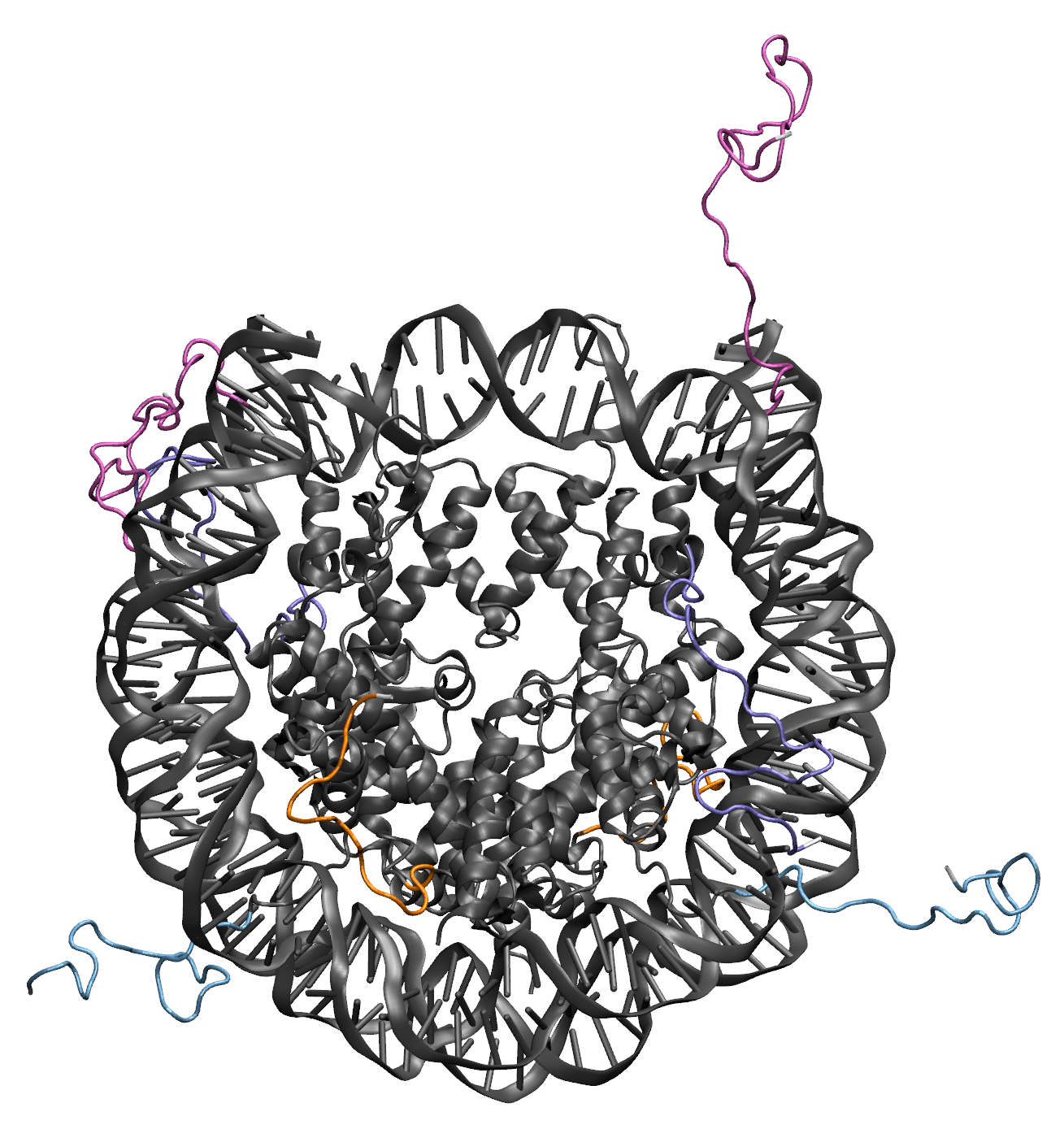
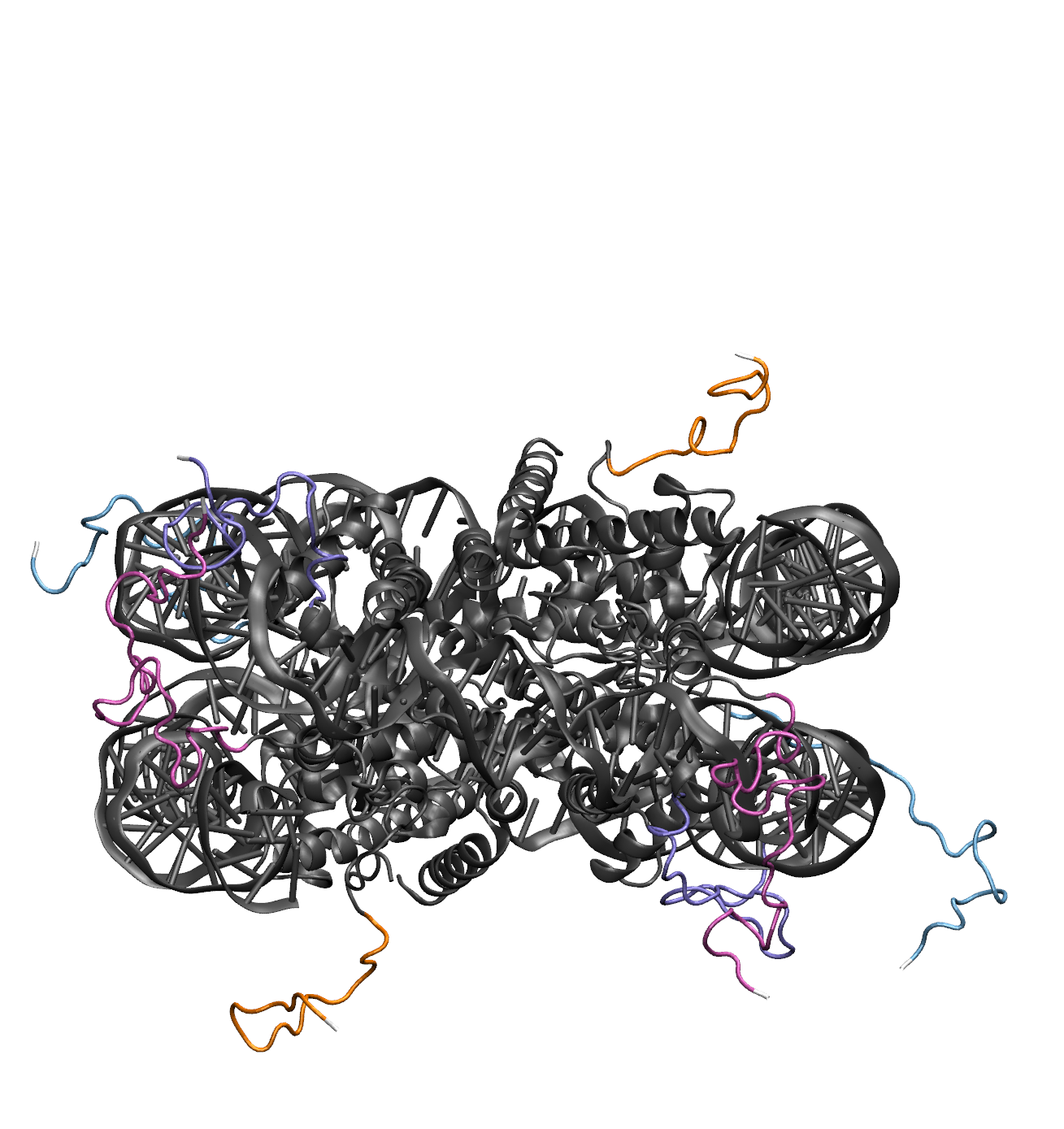 Illustrations of the starting structure highlighting the flexible regions of the H2A proteins in orange, the H2B proteins in teal, the H3 proteins in pink, the H4 proteins in purple, and all post regions in white (note that the move direction dropdown menu is used to set the directionality for defining the post regions for protein). Notice that the terminal residues (white) are not designated as flexible. This prevent problems in the Monte Carlo Sampling caused by the uniqueness of the end residues, which are only connected to one other residue. The input definitions for this selection is as follows:
Illustrations of the starting structure highlighting the flexible regions of the H2A proteins in orange, the H2B proteins in teal, the H3 proteins in pink, the H4 proteins in purple, and all post regions in white (note that the move direction dropdown menu is used to set the directionality for defining the post regions for protein). Notice that the terminal residues (white) are not designated as flexible. This prevent problems in the Monte Carlo Sampling caused by the uniqueness of the end residues, which are only connected to one other residue. The input definitions for this selection is as follows:
move type:
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
move direction:
reverse
reverse
reverse
reverse
reverse
reverse
reverse
reverse
Notice: protein moves use the move direction to designate if the post region should be the resids larger, forward, or smaller, reverse, than those in the flexible region.
flexible regions in order:
segname 1H2A and resid < 20 and resid > 1
segname 2H2A and resid < 20 and resid > 1
segname 1H2B and resid < 27 and resid > 1
segname 2H2B and resid < 27 and resid > 1
segname 1H3 and resid < 40 and resid > 1
segname 2H3 and resid < 40 and resid > 1
segname 1H4 and resid < 31 and resid > 1
segname 2H4 and resid < 31 and resid > 1
post regions in order:
segname 1H2A and resid <= 1
segname 2H2A and resid <= 1
segname 1H2B and resid <= 1
segname 2H2B and resid <= 1
segname 1H3 and resid <= 1
segname 2H3 and resid <= 1
segname 1H4 and resid <= 1
segname 2H4 and resid <= 1
max theta:
30.0
30.0
30.0
30.0
30.0
30.0
30.0
30.0

The output will indicate the location of the output files, acceptance and overlap statistics, and the file names of the inputs, log, and output DCD. Results are written to a new directory within the given "run name" as noted in the output. In addition, a plot of Rg versus structure number is shown (currently NOT implemented).
Several files are generated and saved to the <run name>/monte_carlo/ directory: a copy of the original input PDB and PSF files, the output DCD file containing accepted structures, a PDB and PSF file for each group, flexible region, and post region, the json inputs, and a log file. In this example, the dcd containing the generated structures accepted by the Monte Carlo algorithm is eight_protein_flex/monte_carlo/c36_w601_ncp_eight_protein.dcd.
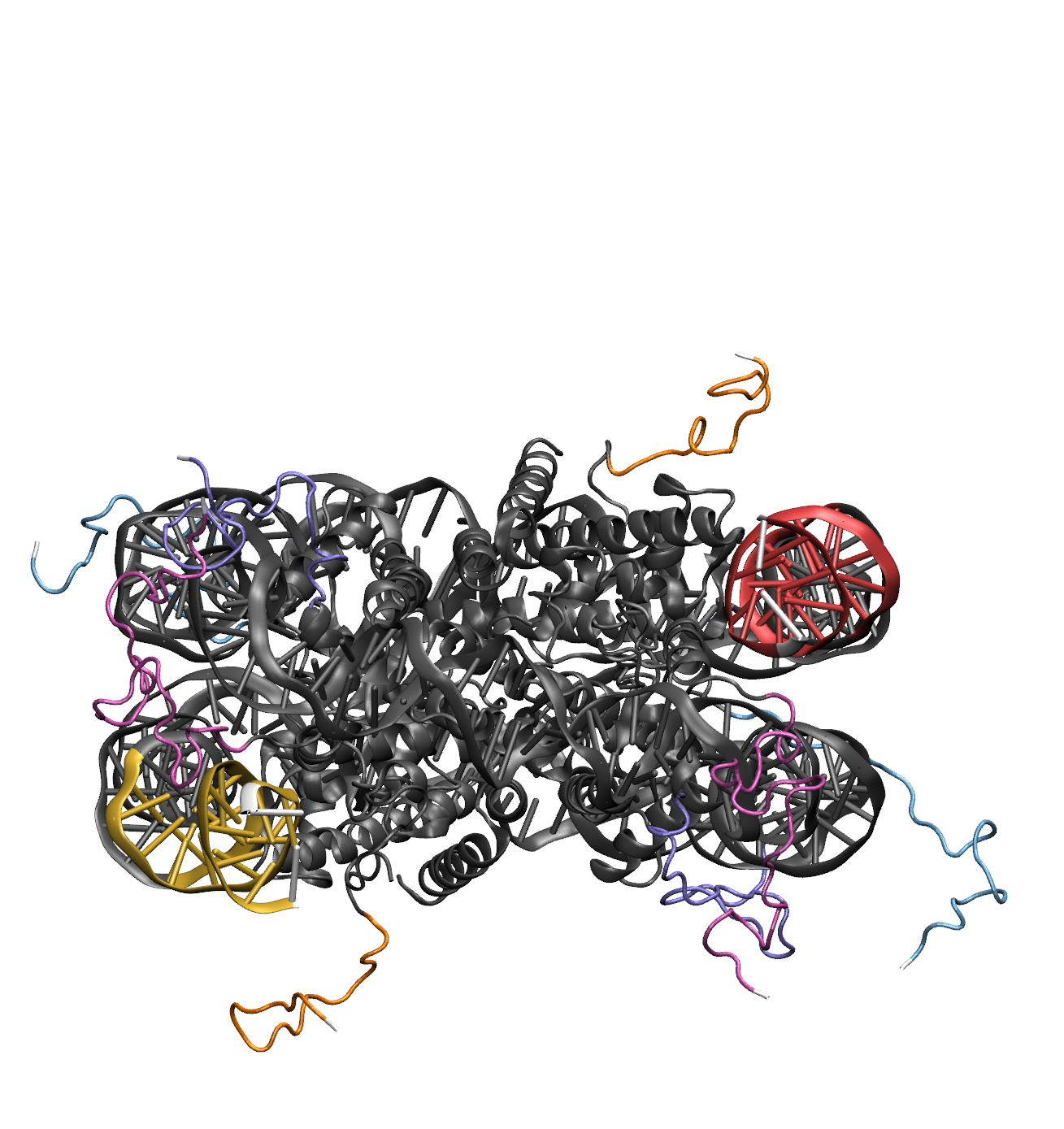 Illustrations of the starting structure highlighting the flexible DNA regions in yellow and red, the flexible protein regions in , and all the post regions in white (note that the directionality used to define the post regions for DNA follows the 5' to 3' direction of the first DNA segment in the flexible region while the protein uses the move direction dropdown menu). The input definitions for this selection is as follows:
Illustrations of the starting structure highlighting the flexible DNA regions in yellow and red, the flexible protein regions in , and all the post regions in white (note that the directionality used to define the post regions for DNA follows the 5' to 3' direction of the first DNA segment in the flexible region while the protein uses the move direction dropdown menu). The input definitions for this selection is as follows:
move type:
double stranded nucleic acid worm-like chain torsion
double stranded nucleic acid worm-like chain torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
protein backbone torsion
move direction:
N/A
N/A
reverse
reverse
reverse
reverse
reverse
reverse
reverse
reverse
flexible regions in order:
(segname DNA1 and resid >= 151 and resid <= 160) or (segname DNA2 and resid >= 194 and resid <= 203)
(segname DNA2 and resid >= 329 and resid <= 338) or (segname DNA1 and resid >= 16 and resid <= 25)
segname 1H2A and resid < 20 and resid > 1
segname 2H2A and resid < 20 and resid > 1
segname 1H2B and resid < 27 and resid > 1
segname 2H2B and resid < 27 and resid > 1
segname 1H3 and resid < 40 and resid > 1
segname 2H3 and resid < 40 and resid > 1
segname 1H4 and resid < 31 and resid > 1
segname 2H4 and resid < 31 and resid > 1
post regions in order:
(segname DNA1 and resid 161) or (segname DNA2 and resid 193)
(segname DNA2 and resid 339) or (segname DNA1 and resid 15)
segname 1H2A and resid <= 1
segname 2H2A and resid <= 1
segname 1H2B and resid <= 1
segname 2H2B and resid <= 1
segname 1H3 and resid <= 1
segname 2H3 and resid <= 1
segname 1H4 and resid <= 1
segname 2H4 and resid <= 1
max theta in order:
10.0
10.0
30.0
30.0
30.0
30.0
30.0
30.0
30.0
30.0

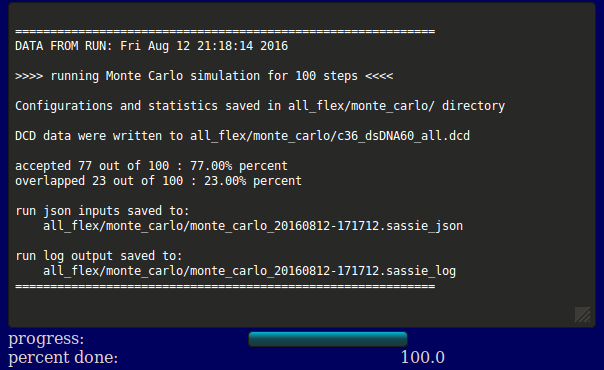
The output will indicate the location of the output files, acceptance and overlap statistics, and the file names of the inputs, log, and output DCD. Results are written to a new directory within the given "run name" as noted in the output. In addition, a plot of Rg versus structure number is shown (currently NOT implemented).
Several files are generated and saved to the <run name>/monte_carlo/ directory: a copy of the original input PDB and PSF files, the output DCD file containing accepted structures, a PDB and PSF file for each group, flexible region, and post region, the json inputs, and a log file. In this example, the dcd containing the generated structures accepted by the Monte Carlo algorithm is all_flex/monte_carlo/c36_w601_all.dcd.
input files
output files
c36_w601_ncp_one_dna.dcd
c36_w601_ncp_two_dna.dcd
c36_w601_ncp_eight_protein.dcd
c36_w601_ncp_all.dcd
Notice that for all of these examples, the end DNA base pair and protein residue was excluded from the flexible region. This prevent problems in the Monte Carlo Sampling caused by the uniqueness of the end bases/residue which only have half the normal connections.
| Protein Backbone | B-DNA | Single-Stranded Nucleic Acid Backbone | Isopeptide Bond | |
|---|---|---|---|---|
| HIV-1 Gag Matrix Protein | X | |||
| Full HIV-1 Gag Protein | X | |||
| Diubiquitin | X | |||
| rpoS mRNA | X | |||
| Linear strand of B-DNA | X | |||
| Nucleosome Core Particle | X | X | ||
| Tetranucleosome | X | X |